

By default, the NBO program is instructed to find the best possible Lewis structure to represent the molecular wavefunction. By "Lewis structure" is meant the pattern of localized 1-center (nonbonded) and 2-center (bonded) electron pairs, corresponding to the familiar Lewis dot diagram. By "best possible" is meant the set of Lewis-type (core, lone pair, bond) NBOs that describe the highest possible percentage of the total electron density. The NBO program considers all possible ways of drawing the bonds and lone pairs of candidate Lewis structure diagrams, as well as the mathematically optimal way of choosing the hybridizations and polarization coefficients in each NBO. The final result of the NBO search is reported in the NBO output section and may be termed the Natural Lewis Structure (NLS) representation of the molecule. (To determine idealized NLS properties in the absence of delocalization corrections, see the $DEL Tutorial.)
Of course, most molecular wavefunctions exhibit some degree of "delocalization" that cannot be described in terms of a single localized Lewis structure representation, even the best possible one. This residual delocalization error of the NLS representation can be quantified in terms of the electronic occupancy of non-Lewis ("starred" BD* valence antibond or RY* Rydberg-type) NBOs that complete the span of the basis. Small non-Lewis occupancies indicate that the wavefunction is well localized and well described at the elementary NLS level. Large non-Lewis occupancies indicate that other "resonance" structures are necessary to describe departures from a single Lewis structure (see the natural resonance theory NRT keyword).
We may sometimes wish to compare the default NBO Lewis structure with a chosen alternative Lewis structure. For this purpose we can use a $CHOOSE keylist to specify a particular Lewis bonding pattern, bypassing the usual NBO search. The program will still determine the optimal hybridizations and polarization coefficients of the requested $CHOOSE bonding pattern, allowing fairest possible comparison with the default NBO structure. The non-Lewis occupancy of the $CHOOSE structure can then be compared with that of the default NBO search to assess the relative accuracy of the two descriptions. Use of the $CHOOSE keylist in this manner allows the user to verify that the program has not inadvertently missed the "true" best Lewis structure. It also allows the user to compare the accuracy of competing descriptions to determine unambiguously which Lewis structure diagram best depicts the molecule, and by how much.
In this Tutorial we employ $CHOOSE keylists to illustrate how an optimal Lewis diagram can be chosen for three representative cases: (i) alternative covalent bonding patterns for 2H + C + O; (ii) alternative representations of BH3:NH3 Lewis acid-base adduct; (iii) alternative representations of phosphine oxide (H3PO).
Given a certain configuration of atoms on the potential energy surface, a frequent question is, "What is the best way to draw the bonding?" In many cases two or more Lewis structure are plausible, and the relevant question is the degree to which one structural formula is better than another. We shall illustrate how to address such questions in the NBO framework using $CHOOSE keylists for atoms in an arbitrarily chosen geometry.
Suppose, for example, we consider a triangular arrangement of two H atoms and one O atom, with a carbon atom somewhere in the middle. If the triangle is highly isosceles, with the two H atoms on the short edge, we might expect a bond pattern like that of single-bonded H2 plus triple-bonded carbon monoxide (viz., 1b in Fig. 1). But if the triangle is more nearly equilateral, as shown in Fig. 1, the 'better' pattern is expected to be that for formaldehyde (1a).
For the particular geometry shown in Fig. 1, with equal distances RCX = 1.15 A from C to each apex atom, let us therefore address the specific question: How much better is the double-bonded formaldehyde structure 1a than the triple-bonded H2 + CO structure 1b for describing the interactions in this geometry?
To answer this question (HF/6-31G* level, singlet surface), we construct distinct $CHOOSE keylists for the alternative Lewis structures. The first specifies a formaldehyde-like bonding pattern
$CHOOSE
lone
4 2
end
bond
s 1 2 s 1 3 d 1 4
end
$END
with two lone pairs on atom O(4), single bonds between atoms C(1)-H(2) and C(1)-H(3), and a double bond between C(1) and O(4). The second specifies a bonding pattern for separated molecular hydrogen and carbon monoxide
$CHOOSE
lone
1 1 4 1
end
bond
s 2 3 t 1 4
end
$END
with one lone pair each on C(1) and O(4), a single bond between H(2)-H(3), and a triple bond between C(1) and O(4).
[Complete details of $CHOOSE syntax, including that for 3-center bonds and open-shell species, are given in the NBO Manual. Note that the "lone" and "bond" lists must each be closed with "end" and that the entire $CHOOSE keylist must be closed with "$END".]
A sample G9X input file for the first $CHOOSE list and the geometry of Fig. 1 is shown below:
#HF/6-31G* pop=NBORead
equilateral H2O triangle, equidistant (1.15 A) from central C
0 1
C
H 1 dist
H 1 dist 2 120.
O 1 dist 3 120. 2 180.
dist 1.15
$NBO $END
$CHOOSE
lone
4 2
end
bond
s 1 2 s 1 3 d 1 4
end
$END
The first portion of the NBO search output for this job is shown below:
NATURAL BOND ORBITAL ANALYSIS:
Occupancies Lewis Structure Low High
Occ. ------------------- ----------------- occ occ
Cycle Thresh. Lewis Non-Lewis CR BD 3C LP (L) (NL) Dev
=============================================================================
1(1) 1.90 15.86201 0.13799 2 4 0 2 1 0 0.06
-----------------------------------------------------------------------------
Structure accepted: NBOs selected via the $CHOOSE keylist
--------------------------------------------------------
Core 3.99945 ( 99.986% of 4)
Valence Lewis 11.86257 ( 98.855% of 12)
================== ============================
Total Lewis 15.86201 ( 99.138% of 16)
-----------------------------------------------------
Valence non-Lewis 0.10749 ( 0.672% of 16)
Rydberg non-Lewis 0.03049 ( 0.191% of 16)
================== ============================
Total non-Lewis 0.13799 ( 0.862% of 16)
--------------------------------------------------------
As shown in the output the NBOs were "selected via the $CHOOSE keylist" (which would have been the same as a default NBO search in this case). The specified $CHOOSE structure led to a Lewis occupancy of 15.86201e (99.14% of the total electron density) and non-Lewis occupancy of 0.13799e (0.86%), corresponding to high overall accuracy of the formaldehyde Lewis structure.
The corresponding output for the second $CHOOSE structure shows a much larger non-Lewis occupancy "error":
NATURAL BOND ORBITAL ANALYSIS:
Occupancies Lewis Structure Low High
Occ. ------------------- ----------------- occ occ
Cycle Thresh. Lewis Non-Lewis CR BD 3C LP (L) (NL) Dev
=============================================================================
1(1) 1.90 14.19269 1.80731 2 4 0 2 2 2 0.92
-----------------------------------------------------------------------------
Structure accepted: NBOs selected via the $CHOOSE keylist
--- Apparent excited state configuration ---
The following "inverted" NBO labels reflect the actual hybrid overlap:
NBO 4 has been relabelled BD*
NBO 34 has been relabelled BD
--------------------------------------------------------
Core 3.99944 ( 99.986% of 4)
Valence Lewis 9.98075 ( 83.173% of 12)
================== ============================
Total Lewis 13.98019 ( 87.376% of 16)
-----------------------------------------------------
Valence non-Lewis 1.96348 ( 12.272% of 16)
Rydberg non-Lewis 0.05633 ( 0.352% of 16)
================== ============================
Total non-Lewis 2.01981 ( 12.624% of 16)
--------------------------------------------------------
The initial $CHOOSE-directed search leads to non-Lewis occupancy of 1.80731e for the H2 + CO structure, more than 13 times worse than the formaldehyde structure. Clearly the H2 + CO bonding pattern 1b is grossly inferior to the formaldehyde bonding pattern 1a for describing the electronic distribution in this geometry.
The inappropriateness of the H2 + CO description is further demonstrated when NBO 4, the 2-center H(2)-H(3) "bond", is actually discovered to have antibonding hybrid overlap between the atoms (giving an "Apparent excited-state configuration"). This NBO is therefore re-labelled "BD*" instead of "BD", leading to a final tabulated non-Lewis occupancy of 2.01981e. The occupancy of NBO 4 is only 0.98e and that of NBO 7 (the forced C(1) "lone pair") is 1.23e, both far below the "pair" requirement for a reasonable Lewis structure. These results confirm (as expected) that only the formaldehyde-type Lewis structure diagram is reasonable for the geometry of Fig. 1.
Consider the equilibrium geometry of the BH3NH3 Lewis acid-base adduct, as depicted in Fig. 2 (RHF/6-31G* level):
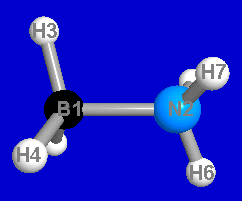
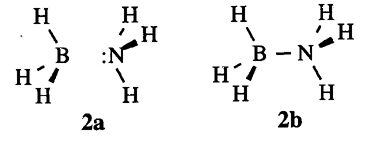
Should this species best be represented as two isolated molecules (BH3 + NH3, 2a) or with a B-N single bond (2b)? If the latter, how does one describe the anomalous "dative" character of the B-N bond?
The two alternative Lewis structures correspond to alternative $CHOOSE keylists
$CHOOSE
lone
2 1
end
bond
s 1 3 s 1 4 s 1 5
s 2 6 s 2 7 s 2 8
end
$END
for the dissociated BH3 + NH3 structure 2a, or
$CHOOSE
bond
s 1 2
s 1 3 s 1 4 s 1 5
s 2 6 s 2 7 s 2 8
end
$END
for the bonded H3B-NH3 structure 2b. The latter is definitely the better representation, with non-Lewis occupancy 0.03734e (0.21% error). The dissociated structure has 10-fold larger non-Lewis occupancy 0.3713e (2.06% error).
The error in the dissociated $CHOOSE structure 2a is almost entirely associated with delocalization from the N lone pair to the unfilled LP* p-type orbital of B. The output describing the corresponding B(1)-N(2) dative bond NBO of the bonded structure 2b is shown below:
(Occupancy) Bond orbital/ Coefficients/ Hybrids
-------------------------------------------------------------------------------
1. (1.99339) BD ( 1) B 1- N 2
( 16.33%) 0.4042* B 1 s( 15.46%)p 5.44( 84.12%)d 0.03( 0.42%)
0.0001 0.3926 0.0208 -0.0004 0.0000
0.0000 0.0000 0.0000 -0.9169 -0.0232
0.0000 0.0000 0.0000 0.0000 0.0651
( 83.67%) 0.9147* N 2 s( 34.52%)p 1.90( 65.47%)d 0.00( 0.00%)
0.0000 0.5873 -0.0173 -0.0006 0.0000
0.0000 0.0000 0.0000 0.8080 -0.0428
0.0000 0.0000 0.0000 0.0000 0.0069
One can recognize that this dative bond NBO is highly polarized toward N, with only 16.33% contribution from the p-rich hybrid on B. If one attempts to describe this electron pair (1.99339e) purely as a lone pair on nitrogen, as in the dissociated $CHOOSE structure 2a, one incurs an error of
which accounts for practically all of the additional non-Lewis occupancy of the inferior structure 2a.
Note that there is no sharp physical boundary separating the limiting case of a highly polarized dative "bond" from a "lone pair." The NBO criterion for a 2-center bond (BD) NBO is that at least 5% contribution must come from each of the two centers. "2-center" NBOs that are more than 95% polarized toward one atom (i.e., with more than 1.90e occupancy on the dominant center) will instead be recognized as a 1-center lone pair (LP). This numerical cut-off (although arbitrary) usually corresponds fairly well to the empirical chemical distinction between a highly polar "bond" vs. a highly delocalized "lone pair."
Let us finally consider the equilibrium geometry of H3PO (phosphine oxide), as shown in Fig. 3 (B3LYP/6-31+G* level):


This is an instructive borderline species that is commonly represented by the double-bonded H3P=O Lewis structure 3b, corresponding to the $CHOOSE structure
$CHOOSE
lone
2 2
end
bond
d 1 2
s 1 3 s 1 4 s 1 5
end
$END
However, default NBO analysis gives the single-bonded Lewis structure 3a, with three lone pairs on O (formal -1 charge on O, +1 on P), corresponding to the $CHOOSE structure
$CHOOSE
lone
2 3
end
bond
s 1 2
s 1 3 s 1 4 s 1 5
end
$END
with total non-Lewis occupancy of 0.47022e (1.81% delocalization error). Each of the two p-type lone pairs on oxygen are strongly delocalized into vicinal P-H* antibonds. (The third, s-rich lone pair is much more localized). The double-bonded structure 3b evidently corresponds to representing one of these two equivalent p-type oxygen orbitals as a lone pair and one as a highly polar "pi bond."
However, it is clearly unphysical to represent only one of the two equivalent p-type oxygen lone pairs in this unsymmetric manner. A more consistent choice is to treat both orbitals as "pi bonds," leading to a $CHOOSE structure as depicted in 3c with formal triple bond between phosphorus and oxygen:
$CHOOSE
lone
2 1
end
bond
t 1 2
s 1 3 s 1 4 s 1 5
end
$END
The table below summarizes the non-Lewis occupancy (NL occ.), %-delocalization error (NL Error), and percentage polarization (%-pol.) of each PO "pi bond" toward phosphorus in the three possible $CHOOSE structures:
| $CHOOSE List | PO bonds | NL Occ.(e) | NL Error(%) | %-pol. | |
|---|---|---|---|---|---|
| structure | 3a | 1 | 0.4702 | 1.81% | [100%] |
| 3b | 2 | 0.6574 | 2.53% | 91.5% | |
| 3c | 3 | 0.8362 | 3.22% | 91.5% | |
This is evidently a borderline case, with two oxygen p-type lone pairs that are so strongly delocalized as to qualify as possible "pi bonds" (only 91.5% on O). Nevertheless, the two pi-bonded structures 3b or 3c clearly have higher overall delocalization errors than the single-bonded structure 3a, which is therefore "best" overall. (Although an argument might be made for a degree of polar pi-bonding in this molecule, structure 3b is quite misleading in this respect and should be replaced by the triple-bonded structure 3c if this aspect of the bonding is to be emphasized.)